在科研的广袤海洋里,单细胞数据的深度解析如精确打击的海战。然而,批次效应这座潜伏在深处的暗礁,若被忽视,将会阻碍科学探索的步伐。此刻,我们汇聚力量,寻找样本整合与批次效应过滤的策略,以保障数据分析的顺畅运行。
一、批次效应:数据分析的隐形杀手
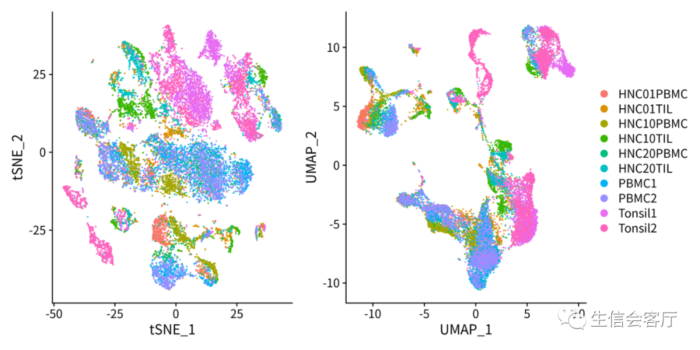
“批次效应”揭示了特定时空条件下数据呈现的全局异质性。由于多台设备采样时间和实验室环境等因素的影响,数据具有各自独特的特性,如同源自多个独立来源。如果不进行适当处理,可能会导致数据分析的混乱,如图所示,将大量单细胞数据简单拼接后,样本间的显著差异使得数据分析变得困难。
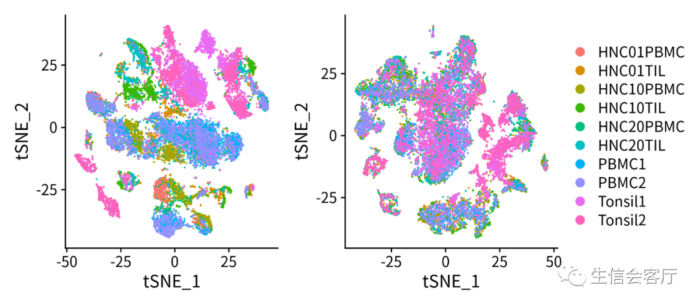
为解决此问题,我司推荐使用”校正批量效应”技术。通过图形展示,可见处理后的样本间协同性能大幅提升,客户业务数据得以回归正常运转轨道,并展现无限潜能。
二、算法大比拼:谁是批次效应的克星?
在单细胞研究领域中,如MNN、CCAMNN、Harmony、Scanorama及scMerge等修正批效应的策略均各具优势。其命名则应用了军事术语,使人自然地联想至勇敢善战的军队,意欲全力应击批效应这一强劲挑战。
CCAMNN因其优越性能及广泛适用性,荣获最新《CELL》期刊专文赞誉,无疑展示了其深厚的技术功底与应用价值。通过Seurat工具箱,使用者能够便捷地运用CCAMNN对大量数据进行校正与跨平台整理,如整合10x、BD、SMART等单细胞数据,形成一股集结各大星系冒险者共同探索宇宙奥秘的力量。
三、CCA分析:降维打击,一击必中
MCA分析,作为一种先进的降维手段,能够将多组独立数据样本的特性映射到低维空间,以此深化对各个因素之间关系的理解及分析。此过程如同跨越浩瀚无垠的宇宙,在同一尺度上的深度交互。
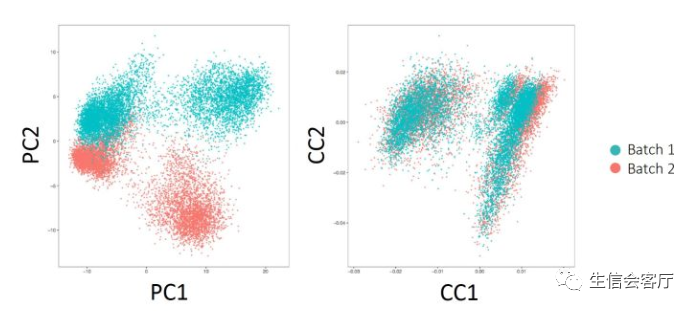
通过CCA降维处理,坐标空间中细胞间距不再代表相似度,反而突显了它们紧密的内在关联。这种特性使性质和状态类似的细胞有可能突破技术限制,形成紧密的聚集态。如图所示,在CCA降维后,细胞间的距离实际反映的是其转录特征的相关性。因此,即使存在技术差异,性质和状态相似的细胞仍能紧密结合,犹如来自不同星系的生物在同一舞台上共舞。
四、MNN算法:寻找最近的邻居
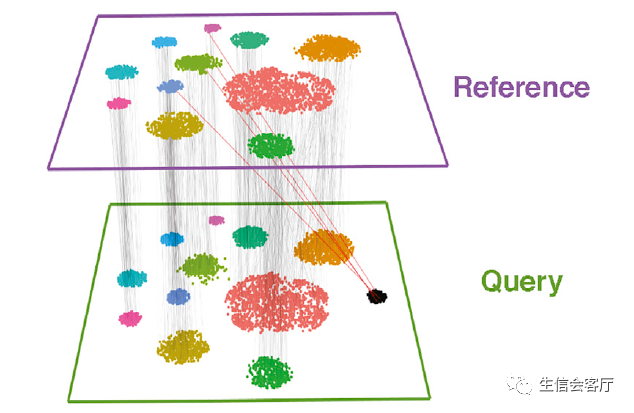
借助CCA降维法,低维度空间内的细胞展现出显著的测距特征。在此基础上,采用模板匹配算法进行细胞识别,效果显著。该算法旨在找出两组数据中距离最短的细胞,将其定义为“锚定细胞”,如同构建星际间的桥梁,实现细胞间的信息传递和学习。
首选方案是从具有类似特性的细胞群体中选择参照依据。然而,部分样本中这类细胞的缺失导致我们发现了其他可行的匹配技法,以颜色较深的红线进行标记。对于从CCA降维空间筛选出来的初选细胞,需要再次回顾核查原始高维基因表达数据。如果这些细胞的转录特性具有高度的相似性,则需保留下来;倘若差异较大,需要进行去除。这个过程就像将来自不同星球的客人邀请到同一场晚会上,当舞步相互配合时即可共同跳舞;而如果差异过大,他们可能需要寻找更适合自己的舞伴。
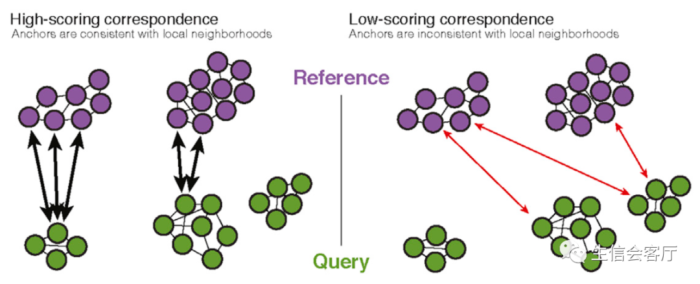
五、锚点可靠性:质量的保证
由左端锚定点引发的多重精确匹配于参照数据中产生了较高信任度得分;然而,仅有右端锚定点引发的单一有限限定匹配却导致信任度评级相对较低。
六、校正后的基因表达值:技术的胜利
精良校正及消弭偏颇数据误差的技巧,使得两个独立单细胞数据能够紧密结合,好似在浩渺宇宙中共赴探索未知的奥秘。然而,并非所有单细胞模型皆需融合,同样重要的是检验整合算法减少技术偏差的能力,防范过度调节导致真实细胞内基因表达变化被忽视。面对这些挑战犹如大洋中的风浪,我们需要用智慧与毅力去克服。
library(Seurat)
library(tidyverse)
library(patchwork)
dir.create('cluster1')
dir.create('cluster2')
dir.create('cluster3')
set.seed(123) #设置随机数种子,使结果可重复
##==合并数据集==##
##使用目录向量合并
dir = c('data/GSE139324_HNC/GSM4138110',
'data/GSE139324_HNC/GSM4138111',
'data/GSE139324_HNC/GSM4138128',
'data/GSE139324_HNC/GSM4138129',
'data/GSE139324_HNC/GSM4138148',
'data/GSE139324_HNC/GSM4138149',
'data/GSE139324_HNC/GSM4138162',
'data/GSE139324_HNC/GSM4138163',
'data/GSE139324_HNC/GSM4138168',
'data/GSE139324_HNC/GSM4138169')
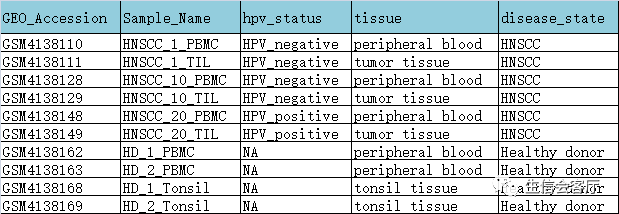
names(dir) = c('HNC01PBMC', 'HNC01TIL', 'HNC10PBMC', 'HNC10TIL', 'HNC20PBMC',
'HNC20TIL', 'PBMC1', 'PBMC2', 'Tonsil1', 'Tonsil2')
counts <- Read10X(data.dir = dir)
scRNA1 = CreateSeuratObject(counts, min.cells=1)
dim(scRNA1) #查看基因数和细胞总数
#[1] 23603 19750
table(scRNA1@meta.data$orig.ident) #查看每个样本的细胞数
#HNC01PBMC HNC01TIL HNC10PBMC HNC10TIL HNC20PBMC HNC20TIL PBMC1 PBMC2 Tonsil1 Tonsil2
# 1725 1298 1750 1384 1530 1148 2445 2436 3325 2709
#使用merge函数合并seurat对象
scRNAlist <- list()
#以下代码会把每个样本的数据创建一个seurat对象,并存放到列表scRNAlist里
for(i in 1:length(dir)){
counts <- Read10X(data.dir = dir[i])
scRNAlist[[i]] <- CreateSeuratObject(counts, min.cells=1)
}
#使用merge函数讲10个seurat对象合并成一个seurat对象
scRNA2 <- merge(scRNAlist[[1]], y=c(scRNAlist[[2]], scRNAlist[[3]],
scRNAlist[[4]], scRNAlist[[5]], scRNAlist[[6]], scRNAlist[[7]],
scRNAlist[[8]], scRNAlist[[9]], scRNAlist[[10]]))
#dim(scRNA2)
[1] 23603 19750
table(scRNA2@meta.data$orig.ident)
#HNC01PBMC HNC01TIL HNC10PBMC HNC10TIL HNC20PBMC HNC20TIL PBMC1 PBMC2 Tonsil1 Tonsil2
# 1725 1298 1750 1384 1530 1148 2445 2436 3325 2709
七、SingleR:人工鉴定的可靠方法
深度探索深度信息中dim与table功能之后,尽管两种方法截然不同,我们依然取得了基因与细胞数相同的结论,无可辩驳地证实了”SingleRMarker基因人工鉴定”在细胞分类精确度以及可靠性上的出色表现。同时,以两套标准数据库为例进行的SingleR实验数据也进一步支持这一观点,尽管存在微小差异,但无疑再次肯定了上述结论的准确性。
scRNA1 <- NormalizeData(scRNA1)
scRNA1 <- FindVariableFeatures(scRNA1, selection.method = "vst")
scRNA1 <- ScaleData(scRNA1, features = VariableFeatures(scRNA1))
scRNA1 <- RunPCA(scRNA1, features = VariableFeatures(scRNA1))
plot1 <- DimPlot(scRNA1, reduction = "pca", group.by="orig.ident")
plot2 <- ElbowPlot(scRNA1, ndims=30, reduction="pca")
plotc <- plot1 plot2
ggsave("cluster1/pca.png", plot = plotc, width = 8, height = 4)
print(c("请选择哪些pc轴用于后续分析?示例如下:","pc.num=1:15"))
#选取主成分
pc.num=1:30
##细胞聚类
scRNA1 <- FindNeighbors(scRNA1, dims = pc.num)
scRNA1 <- FindClusters(scRNA1, resolution = 0.5)
table(scRNA1@meta.data$seurat_clusters)
metadata <- scRNA1@meta.data
cell_cluster <- data.frame(cell_ID=rownames(metadata), cluster_ID=metadata$seurat_clusters)
write.csv(cell_cluster,'cluster1/cell_cluster.csv',row.names = F)
##非线性降维
#tSNE
scRNA1 = RunTSNE(scRNA1, dims = pc.num)
embed_tsne <- Embeddings(scRNA1, 'tsne') #提取tsne图坐标
write.csv(embed_tsne,'cluster1/embed_tsne.csv')
#group_by_cluster
plot1 = DimPlot(scRNA1, reduction = "tsne", label=T)
ggsave("cluster1/tSNE.png", plot = plot1, width = 8, height = 7)
#group_by_sample
plot2 = DimPlot(scRNA1, reduction = "tsne", group.by='orig.ident')
ggsave("cluster1/tSNE_sample.png", plot = plot2, width = 8, height = 7)
#combinate
plotc <- plot1 plot2
ggsave("cluster1/tSNE_cluster_sample.png", plot = plotc, width = 10, height = 5)
#UMAP
scRNA1 <- RunUMAP(scRNA1, dims = pc.num)
embed_umap <- Embeddings(scRNA1, 'umap') #提取umap图坐标
write.csv(embed_umap,'cluster1/embed_umap.csv')
#group_by_cluster
plot3 = DimPlot(scRNA1, reduction = "umap", label=T)
ggsave("cluster1/UMAP.png", plot = plot3, width = 8, height = 7)
#group_by_sample
plot4 = DimPlot(scRNA1, reduction = "umap", group.by='orig.ident')
ggsave("cluster1/UMAP.png", plot = plot4, width = 8, height = 7)
#combinate
plotc <- plot3 plot4
ggsave("cluster1/UMAP_cluster_sample.png", plot = plotc, width = 10, height = 5)
#合并tSNE与UMAP
plotc <- plot2 plot4 plot_layout(guides = 'collect')
ggsave("cluster1/tSNE_UMAP.png", plot = plotc, width = 10, height = 5)
##scRNA2对象的降维聚类参考scRNA1的代码
面对科研挑战,我们诚挚邀请您共同努力,确保数据分析的流畅与准确,同时挖掘未来发展可能。